Approach
-
Group theoretical methods and in-house derivative structural algorithms (e.g. SOCCR) are used to enumerate possible anion orderings in candidate structures
-
We perform density functional theory calculations to investigate the atomic and electronic structure of heteroanionic materials. We perform these calculations with the following goals:
- Aiding synthetic collaborators through phase stability calculations in solid state and aqueous conditions.
- Discovering the origin of structure-property relationships specific to heteroanionic materials.
Selected Publications
Structural Diversity from Anion Order in Heteroanionic Materials
Heteroanionic materials leverage the advantages offered by two different anions coordinating the same or different cations to realize unanticipated or enhanced electronic, optical, and magnetic responses. Beyond chemical variations offered by the anions, the ability to control the anion order present within a single transition metal polyhedron via anion- sublattice engineering offers a potentially transformative strategy in tuning material properties. The set of design rules for realizing and controlling anion order, however, are incomplete, which is due in part to the limited anion-ordered diversity in known structures. This aspect makes formulating such principles from experiment alone challenging. Here, we demonstrate how computational methods at multiple levels of theory are effective at investigating the anion site order dependencies in heteroanionic materials, HAMs, and enable the construction of crystal- chemistry principles. Our approach relies on a database of anion ordered structure variants in which we manipulate the lattice degrees of freedom through the incorporation of structural distortions. Structure−property relationships and anion-order descriptors are data mined from group theoretical techniques and density functional theory calculations. Using our combined computational scheme, we uncover a hybrid improper mechanism to stabilize polar phases, propose the chemical link between local and long ranger anion order, and detail the sequence of order−disorder/displacive transitions observed experimentally in the oxyfluoride Na3MoO3F3. Our method is scalable and transferable to many heteroanionic chemistries and crystal families, facilitating the construction of heteroanionic materials design principles.
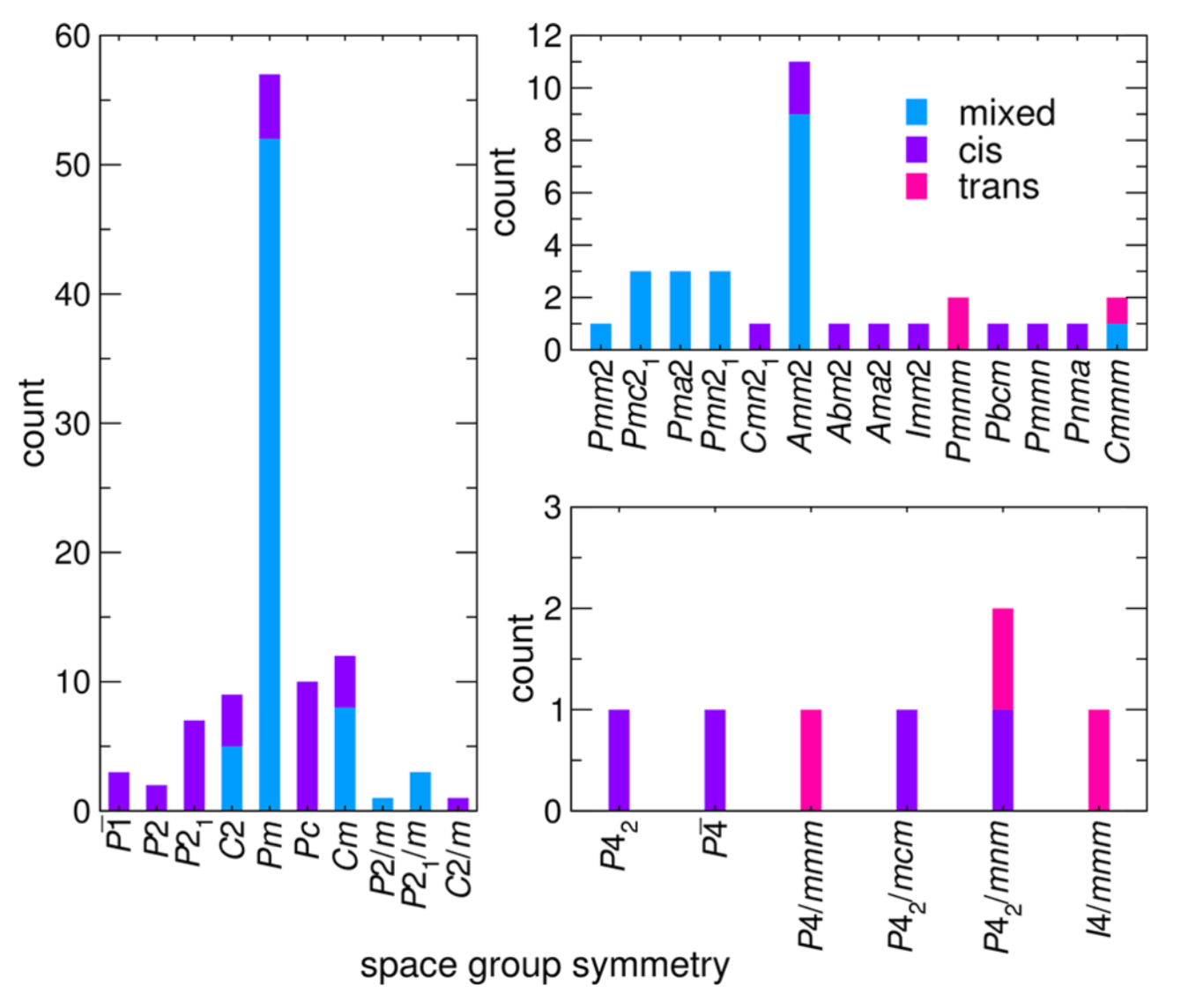
Figure: Occurrence of A2BMO2F4 derivative structures with cis, trans, and mixed isomer configurations sorted by space group symmetry.
Reconstructive Transitions from Rotations of Rigid Heteroanionic Polyhedra
Phase transitions are ubiquitous in structurally complex transition metal compounds composed of homoanionic polyhedra, including nitrides, oxides, and fluorides. The symmetry breaking that occurs across polymorphic transitions is often achieved by small atomic displacements, rendering these displacive transitions reversible. In contrast, elemental crystals, alloys, and simple minerals will exhibit reconstructive “bond-breaking” transitions. Here we show that a reconstructive transition occurs in the heteroanionic compound KNaNbOF5, owing to reorientations of the [NbOF5]2− units that trigger a reconfiguration of the cation lattice. Using a combination of synchrotron-based measurements, empirical dynamic simulations, and ab initio calculations, we report structure changes across the transition and formulate an atomistic minimum energy transition path to explain its irreversible nature. Our results indicate that multianionic compounds are likely to host reconstructive transitions that are frequently difficult to study and functionalize in simpler compounds. We anticipate that our insight into the forces that drive the transition will also lead to novel methods to control the assembly of structures in the solid state.
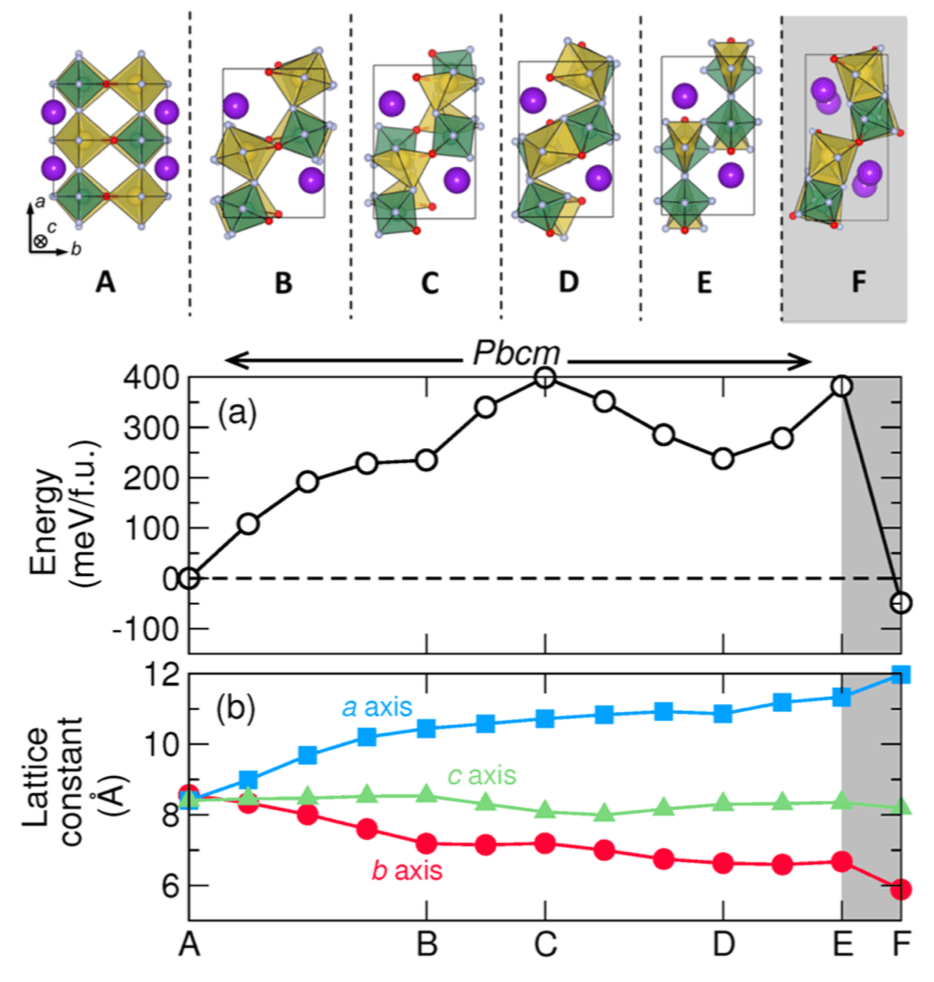
Figure: Properties and structure along the minimum energy phase transformation path at zero pressure. The reaction coordinate is accumulated configurational change along the path. Here the letters indicate key structures, shown above. The region shaded gray represents the reversible HT-to-NCS transition not investigated by the G-SSNEB.