Materials at the Edge
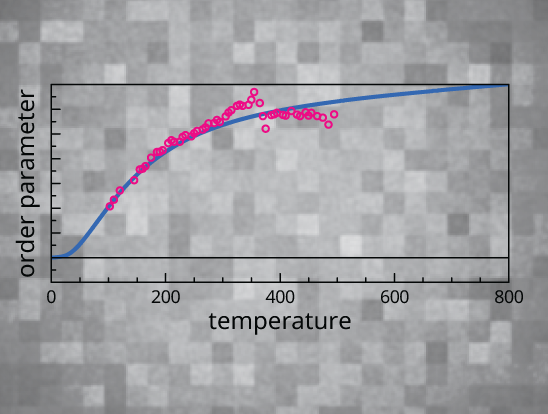
We aim to understand material functionality near phase boundaries by correlating properties to structural building blocks.
Structure Directing Forces
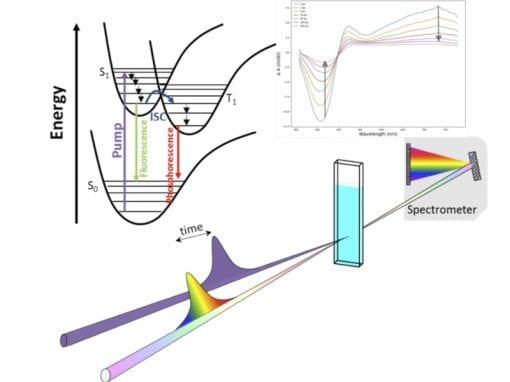
We seek to formulate reliable “bottom-up” design strategies to tune local and crystal symmetry.
Multifunctional Materials Design
We overcome contraindicated materials properties – those that often do not coexist – and rationally unite them in novel structures.
Approach
Our approaches involve harnessing quantum-mechanical approaches, such as density functional theory (DFT) methods, applied group (representation) theory, symmetry analysis, and data analytics (informatics) methods with phenomenological models and crystal-chemistry principles to establish structure–property relationships that can be used for materials design. An advantage of the DFT methods is that they are capable of disentangling the roles of many degrees of freedom (DOF) in determining material function. DFT can predict the spatial distribution of electrons – the electronic charge density – and the corresponding orbital energies.
The ground state properties of a material are obtained through calculation of its electron density, ρ(r), which uniquely defines the energy, E, of the system: E[ρ(r)] = F[ρ(r)] + ∫dr Vext(r)ρ(r). Here F[ρ(r)] is a universal functional describing the internal quantum mechanical interactions of the electrons, and ∫dr Vext(r)ρ(r) is the external potential acting on the electrons from the nuclei and any external fields. In other words, all of the quantum mechanical (term 1) and the atomic structures details (term 2) are treated on equal footing.
This information allows us to explore how bonding between atoms occurs and subsequently predict properties that originate from these interactions. These calculations are also remarkably powerful tools for multi-property design based on structure-driven models; when leveraged with experimental input, they become indispensable to the discovery of new materials.
Browse among our various research themes